C’est une lueur d’espoir pour des milliers de familles confrontées à l’une des formes d’épilepsie les plus cruelles et les plus complexes. Un nouveau médicament, le zorevunersen, vient de livrer des résultats spectaculaires lors d’essais cliniques préliminaires, réduisant la fréquence des crises jusqu’à 91 % chez certains jeunes patients. Contrairement aux traitements classiques qui ne font que masquer les symptômes, cette molécule révolutionnaire s’attaque directement à la mutation génétique responsable de la maladie. Si ces données se confirment, nous pourrions être face au premier traitement capable de modifier radicalement l’évolution du syndrome de Dravet et d’améliorer durablement le développement des enfants.
S’attaquer à la racine du mal : le gène SCN1A
Le syndrome de Dravet ne se limite pas à des convulsions violentes et imprévisibles. Les enfants touchés souffrent également de retards de développement neurologique, de troubles de la coordination et de graves problèmes comportementaux. Plus terrifiant encore, environ la moitié des patients décèdent subitement et prématurément des suites de cette pathologie. À l’origine de ce calvaire se trouve une mutation du gène SCN1A, qui empêche la production normale d’une protéine essentielle à la communication entre les neurones.
Jusqu’à présent, les médicaments antiépileptiques traditionnels et les implants pouvaient réduire la fréquence des crises, mais ils restaient impuissants face aux retards de développement. Le zorevunersen change la donne. Il s’agit d’un oligonucléotide antisens conçu pour « booster » la copie saine du gène SCN1A afin de compenser celle qui est défaillante. En augmentant la production d’ARN messager, le médicament restaure la signalisation des interneurones et corrige le problème à sa source biologique.
Une efficacité foudroyante observée en clinique
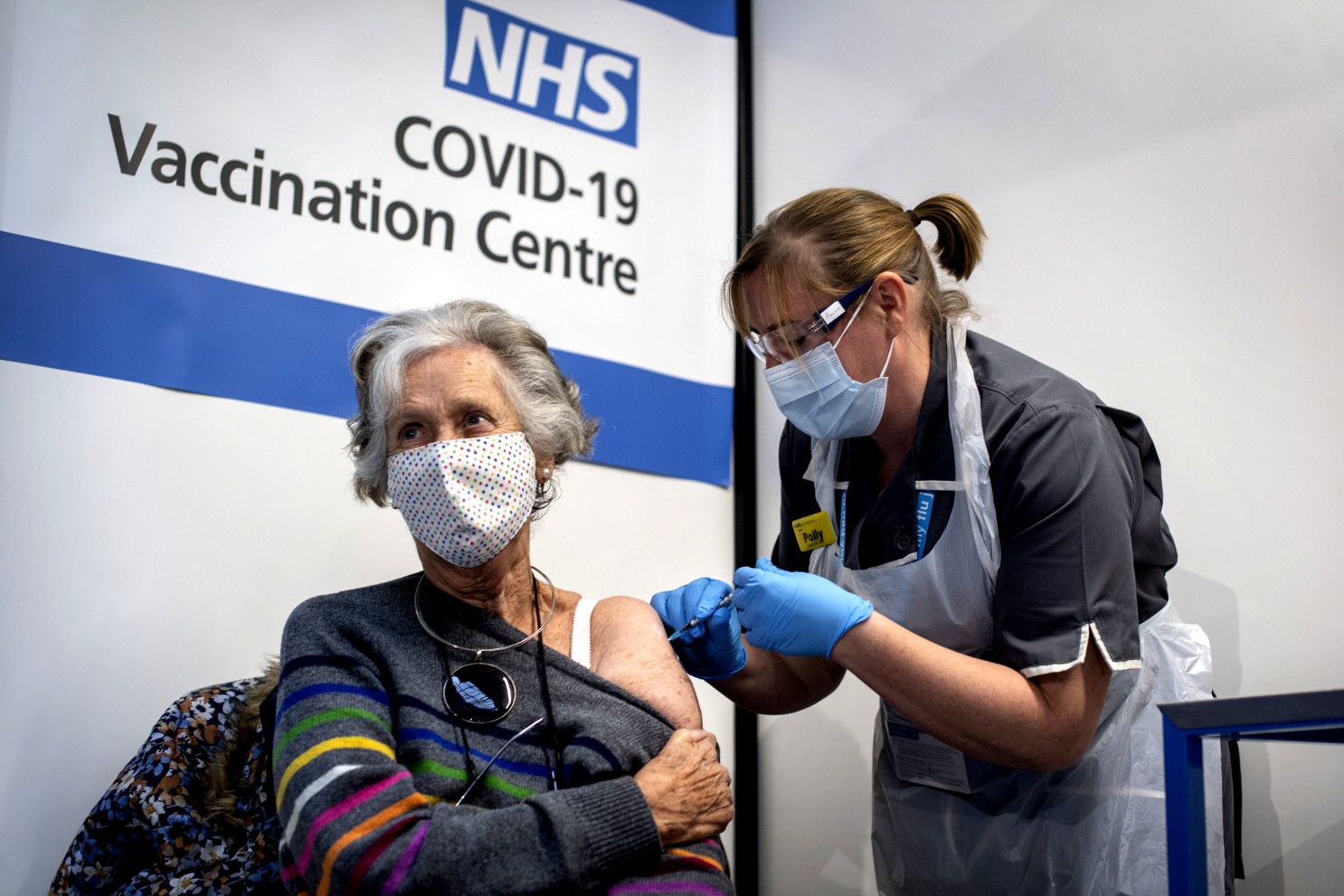
L’étude, publiée le 4 mars 2026 dans le prestigieux New England Journal of Medicine, a suivi 81 enfants âgés de 2 à 18 ans pendant trois ans au Royaume-Uni et aux États-Unis. Pour garantir que le médicament atteigne directement le cerveau, il a été administré par ponction lombaire, une injection dans la colonne vertébrale permettant au produit de se diffuser dans le liquide céphalo-rachidien.
Bien que l’objectif principal fût de tester la sécurité du produit, les bénéfices observés ont dépassé les espérances des chercheurs :
Réduction massive des crises : Après 20 mois de traitement, les enfants ayant reçu les doses les plus élevées ont présenté entre 59 % et 91 % de crises en moins.
Impact sur le développement : L’équipe a noté des améliorations significatives dans le développement neurologique et la coordination motrice.
Qualité de vie : Les participants ont montré une meilleure qualité de vie globale, les effets d’une seule dose pouvant durer plusieurs mois.
Crédit : Moment Makers Group/istock
Sécurité et limites d’une phase précoce
Sur le plan de la tolérance, l’essai s’est révélé rassurant. Si quelques effets secondaires légers comme des maux de tête, des vomissements ou une hausse du taux de protéines dans le liquide céphalo-rachidien ont été notés suite aux injections, le médicament a été jugé sûr pour une utilisation pédiatrique.
Toutefois, les scientifiques appellent à la prudence. Cette étude préliminaire ne comportait pas de groupe placebo et portait sur un échantillon restreint de patients. « Il s’agit de l’un des premiers essais visant à modifier l’évolution de l’épilepsie complexe à début précoce », explique le Dr Helen Cross, responsable de l’étude à l’University College London. L’enthousiasme est réel, mais la validation scientifique complète nécessite une étape supplémentaire.
Horizon 2028 : l’attente d’une validation mondiale
Pour confirmer ces résultats historiques, un essai clinique de plus grande envergure est déjà en cours. Il mobilise 170 enfants supplémentaires afin de comparer rigoureusement l’efficacité du zorevunersen face à un groupe témoin. Cette étape cruciale permettra de déterminer si l’amélioration est statistiquement supérieure à celle d’un traitement standard.
L’essai devrait s’achever officiellement en octobre 2028. Si les conclusions sont positives, il faudra encore quelques années de procédures administratives avant que ce traitement ne devienne accessible à l’ensemble des enfants atteints du syndrome de Dravet. Pour les familles, l’enjeu est immense : passer d’une gestion de crise permanente à une véritable réparation de la cause profonde de la maladie.
Brice est un journaliste passionné de sciences. Il collabore avec Sciencepost depuis plus d'une décennie, partageant avec vous les nouvelles découvertes et les dossiers les plus intéressants.

 3 month_ago
120
3 month_ago
120
 Crédit : Moment Makers Group/istock
Crédit : Moment Makers Group/istock





















.jpg)






 French (CA)
French (CA)